 切换行业
切换行业

背景简介
活性电极材料中的离子插层对许多电化学储能系统来说是极其关键的。层状材料因其超快的插入动力学而特别具有吸引力。MXene是一类快速增长的二维(2D)亲水和分层金属导电材料,在中性和酸性水溶液中表现出了很好的插层电容和动力学。迄今为止,研究最多的MXene,即Ti3C2,在H2SO4水电解质中实现了惊人的体积电容,高达1500 F cm-3,面积电容高达4 F cm-2。通过90 nm厚的Ti3C2或垂直排列的MXene在3M H2SO4水溶液中实现了高达100 V s-1的超高速电容性能,超过了许多其他最先进的电容材料。充电机制显示出对所用电解液的强烈依赖性。
通过原位X射线吸收(XAS)和拉曼光谱分别证明了酸性电解质中的电化学可逆氧化还原(伪电容)机制和中性水介质中的电双层(EDL)响应。Wang等用Ti3C2 MXene在双三氟甲磺酰亚胺锂(LiTFSI)中与不同的有机溶剂进行了假电容Li+互溶,高效地将Li+从碳酸丙烯酯溶剂中解离出来,从而获得了高体积电容和快速电荷传输。然而,不燃不腐、安全、环保、相对高电压、低成本的特点使中性水电解质成为工业上较好的选择。
要增加MXene中储存的能量,就需要更好地理解阳离子的界面化学,包括在限制条件下阳离子,水和MXene表面的相互作用。阳离子嵌入MXene层间可以显著影响层间距、承压水动力学、电导率等。此外,承压水显著提高MXene的能量密度和功率密度。X射线衍射(XRD)、电化学石英晶体微天平(EQCM)和中子散射技术进一步深入了解MXene体积变化的程度及其与共嵌入水含量的关系。
基于电化学膨胀计和原子力显微镜(AFM)的研究结果补充了这一点,这些发现是由于阳离子插入而引起的电极体积和机械刚度的变化。最近的一项报道表明阳离子插层通过XAS显著改变了Ti3C2 MXene中Ti原子的氧化状态。
为了对离子插层过程的有一个基本的了解,通常采用理论与实验相结合的方法。例如,应用ReaxFF-GCMC研究了单层水的扩散系数和阳离子的动力学。Yamada等人利用溶剂化模型对电容进行了研究。虽然这两种方法都与实验结果相吻合,但由于理论参数验证不充分以及缺乏明确的水,预计会导致不正确的溶剂化环境和阳离子MXene相互作用,这对电容性能非常重要,但研究较少。
文章介绍
近日,美国橡树岭国家实验室Nina Balke老师,Weiwei Sun老师等人与其他课题组合作在国际知名期刊Energy & Environmental Science (影响因子:30.289) 上发表题为“Tracking ion intercalation into layered Ti3C2 MXene films across length scales”的研究工作。
本文报告了一个高度集成的研究,其中涉及多尺度理论/模型和实验,以追踪将Li +,Na +,K +,Cs +和Mg2+离子嵌入Ti3C2 MXene中的过程。通过理论/建模辅助的综合实验分析,可以深入了解能量存储过程,从而突出阳离子动力学的重要性,它们在MXene片之间的位置,它们对机械性能和电容性能量存储的影响。
计算模拟和原位量热法测量证明了涉及阳离子脱水和H+再水化的过程,显示了实验和理论之间的热量变化的良好相关性。
原位液态原子力显微镜测绘的离子能量耗散在MXene表面出现非均匀性,表明MXene内部离子的异质性,部分证实了理论中得到的离子行为。
研究者直接证明,当阳离子与MXene表面的平均距离与开路电势电容作图时,遵循一个修正的双面亥姆霍兹模型,揭示了一种不同的电双层禁锢机制。这种新的基本认识为利用二维材料制成的电极和膜改进功能器件奠定了基础。
该文章共同第一作者为Qiang Gao, Weiwei Sun。Nina Balke,Weiwei Sun为本文的通讯作者。
要点解析
要点一:通过MD模拟,探究MXene层间的阳离子排列及其与水的相互作用。
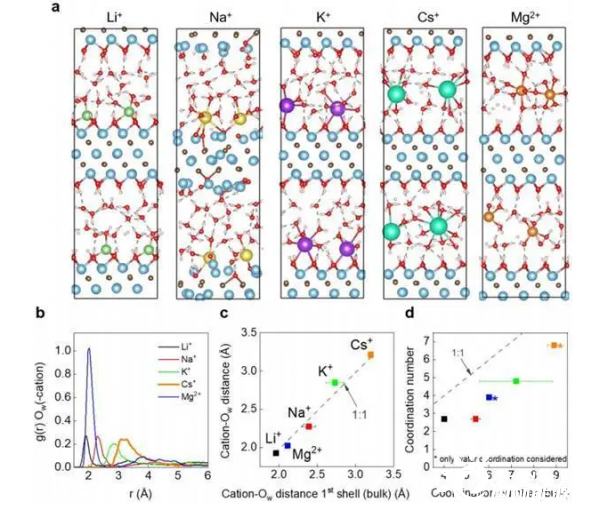
图1.
(a) 层间空间的阳离子分布。浅蓝色代表Ti,深灰色代表C,红色代表O,浅粉色代表H。
(b)根据AIMD建模得到的水中氧的径向分布函数g(r),Ow,围绕阳离子的分布。
(c) 与g(r)的最大值位置相对应的阳离子-Ow距离显示出与散装溶液中的数值有明显的相关性。
(d)从b中提取的配位数与散装溶液的数值比较。取散装溶液的配位数,并对参考文献中提供的所有数值进行平均,灰线表示当密闭CN与散装溶液中的CN相等时,即1:1时的参考。
图1a显示了限制在两个MXene层中的不同阳离子周围的代表性原子排列。每个阳离子对水分子和MXene表面表现出不同的亲和力。Li+,Na+和K+在孔表面显示特定吸附,而Cs+和Mg2+位于孔中心。在所有研究的阳离子中,Na+引起了MXene的无序结构。
如图1b所示,Li +和Mg2+的阳离子-Ow距离最短,即为水合壳半径最小,而Cs+的水合半径最大。
如图1c所示,阳离子-Ow之间的距离与游离水中各自的值成线性比例关系。证明1st水合半径不受限制的影响。
如图1d所示, 与溶剂化壳半径相比,约束阳离子的配位数(CN)与体积值相比明显减少,表明所有阳离子进入MXene的密闭空间后都会发生部分脱水。
要点二:对阳离子/水交换反应的能量学的研究。
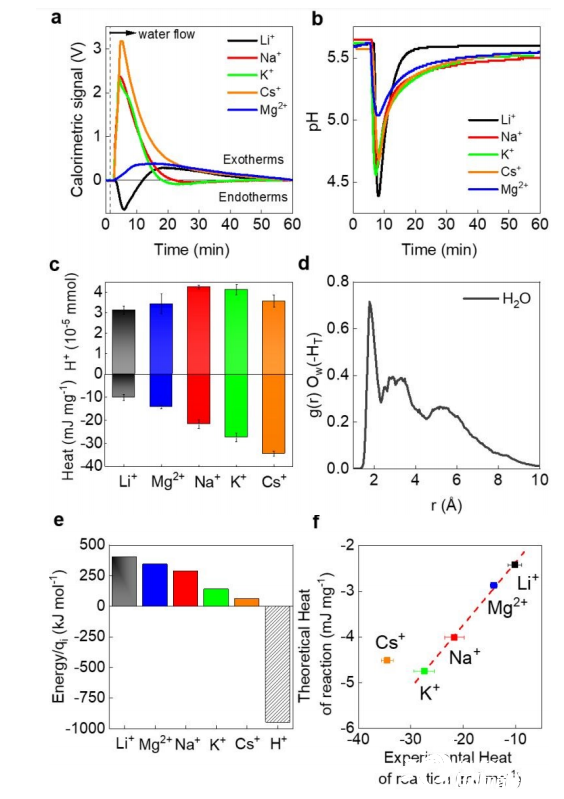
图2.
(a)在水与阳离子交换过程中量热信号的时间依赖性。
(b)在量热法中进行的pH测量。
(c)定量分析由于阳离子吸附到MXene末端的-OH位上而释放的H+和由(a)确定的反应热。
(d)从AIMD模拟中获得的纯水在H周围到终端(HT)的氧在水中的径向分布函数,g(r),Ow。
(e)通过阳离子化合价归一化的阳离子脱水和H +脱水能。
(f)使用AIMD在理论上计算的和使用量热法在实验上测量的之间的反应相关热。
图2a中可见,在~15分钟开始时有持久的内热信号,因此所测得的反应热(HOR)确实与阳离子的原子大小呈现出同步趋势,其中Li+夹杂MXene的反应热调制最大。
测量的pH值随时间的变化如图2b所示。Li+的pH信号比其他阳离子更快地恢复到基线,从而使H+释放量最小。在图2c中,从图2a-b中提取的以mJ mg-1(MXene质量)为单位的HOR和释放的H+量之间没有任何相关性(图2c),但HOR本身呈现出Li+<Mg2+<Na+<K+<Cs+的明显趋势。
利用从g(r)函数中提取的表面质子(HT)的CN(图2d)的内热和放热部分来模拟测量的HOR。
图2e中显示了通过阳离子电荷归一化的相应能量,并且是根据CN相对于全水合能的分数变化计算的。
将测量值与理论模型值进行比较,我们发现实验与理论之间存在定性的线性相关,Cs+为离群值(图2f),但能量尺度相差4.2-7.6倍。
要点三:对限制中阳离子驱动的能量损失的研究
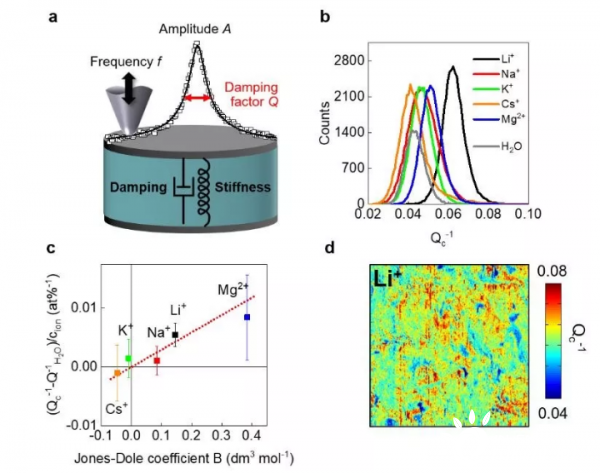
图3.
(a)CR-AFM的示意图。
(b)在机械振动下样品的能量耗散Qc -1的测量分布与电解质的关系。
(c)相对于纯水的能量耗散变化,其通过离子浓度归一化并相对于琼斯·多尔系数B作图。
(d)在浸入水中两个小时记录的0.5 M Li2SO4中测得的局部能量损失的空间图(尺寸10×10 ?m)
为了研究局部的弹性模量和能量耗散过程,采用了横向分辨率为10nm的接触共振原子力显微镜(CR-AFM)。当机械激励时,与表面接触的AFM探针振荡是杨氏模量和能量耗散的函数,用阻尼因子Q-1的倒数表示(图3a)。
实验表明,阳离子类型对MXene的机械耗能有显著的影响(图 3b)
图3c中的误差条表示样品表面的变化,单个像素处的测量不确定度要小得多。图3c显示了通过坐标系原点的拟合线的线性相关性。
如图3d为Li+在10s纳米长度内的离子分布图。结果表明,Li+离子的能量耗散是不均匀的,在样品表面出现了高能量损失的斑块。
要点四:探究电荷储存机制
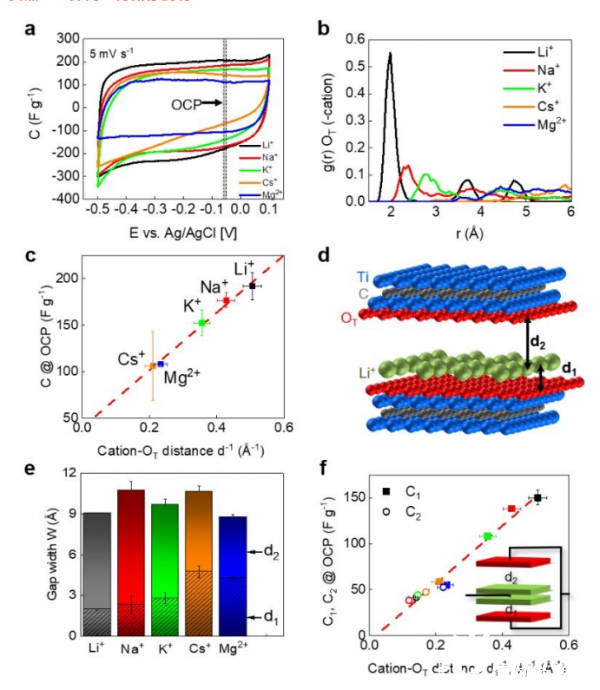
图4.
(a)在0.5 M硫酸盐溶液中不同阳离子的循环伏安图(5 mV s-1)。
(b) MXene表面氧离子的径向分布函数。
(c) d-1对应的阳离子MXene distance-1与OCP处平均电容的关系。
(d) 两个MXene层中改进的Helmholtz模型示意图。d1和d2随水性阳离子而变化。
(e) 根据AIMD模型确定的宽度为W=d1+d2的受限空间。
(f) C1和C2之间的关系,以及OCP处对应的1/d1和1/d2的反距离。插图:得到了一个含有两个并联电容器的等效电路,其间距为d1和d2。
通过循环伏安图(CVs)在5mv s-1下测得的阳离子MXene距离与电容之间的联系(图4a)。矩形CVs表明主要电荷储存机制是EDL。
一旦阳离子插入MXene中,它们占据MXene层的优选位置,这些位置与MXene表面的距离不同,通过作为MXene表面参考点的终端氧(-OT)周围阳离子的RDF进行探测(图4b)。Li+最接近表面,其次是Na+和K+。g(r)中发现的这一系列趋势在质量上与以前基于ReaxFF的研究中的趋势相同。
研究者们发现OCP处的电容与距离d的反阳离子之间的线性关系,截距为39.9±6.6 F g-1(图4c)。
为了合理化剩余截获电容,提出了一个修正的Helmholtz模型,图4d为宽度为W=d1+d2的受限空间,被同一区域A的两个MXene表面所夹。d1是阳离子和MXene表面之间的最近距离
根据AIMD(图4e)确定宽度W后,将总电容分解为从d1和d2(W-d1)导出的C1和C2,以获得模拟的最佳统计数据。当在1/d上单独绘制时,将导出截距为-6.1±5.4 F g-1的线性关系,即拟合穿过图形的原点(图4f)。C2电容对于所有阳离子都具有可比性,集中在40-50fG-1左右,这非常接近图4c中的电容截距,这表明电容截距源于与d2相关的表面电容。
结论
鉴于在限制中电解质和电极之间的相互作用的复杂性质,本文观察到的阳离子和MXene表面之间的相互作用有希望在纳米流体的分层材料中得到更广泛的应用。在强约束下,该体系以阳离子水化和阳离子与表面终止基团的交换(阳离子吸附)为主。 所展示的阳离子-MXene相互作用的水性阳离子对宏观材料行为有影响,如阳离子交换能量,机械能量损失,这可用于研究夹层过程的空间变化和电化学电容。
本文证明了阳离子和MXene表面之间的平均距离遵循1/d的关系。本文提出了修正的赫尔姆霍兹模型来阐明MXene的EDL机制,该模型适用于广泛的水电解质和分层材料。捕获离子和质子水合的合理配位数是理解受限条件下的能量学的关键,这些突出显示了在限制条件下了解阳离子-水-MXene相互作用的重要性,以便全面了解MXenes的储能特性和过程。只有将具有明确描述的水和固液相互作用的框架的实验和从头算理论结合在一起并进行分析以揭示相关关系时,这才有可能。
本文展示的方法和研究结果将增强读者对二维材料储能的理解,并有助于指导MXenes家族的进一步发展。预计综合研究将被迅速采用,从而能够理解和操纵用于电化学储能、海水淡化和离子选择性膜的离子和密闭流体的行为。




 正在加载...
正在加载...


